
بررسیو تشریح عارضه(Hemophagocytic Lympho Histiocytosis (HLH نیاز بهچالش هوشمندانه بهعلت تعریف ناقص این سندرم دارد. زیرا مشابهت و صفات مشترک داشتن(Overlap) با پرزانتاسیون حالات Inflammatory دارد. نامشخصبودن تشخیص چالش عملی را در درمان فوری این افراد که باید داروهای ایمیونوسوپرسیو دریافت دارند میافزاید. درمان موفقیتآمیز زمانی بهدست میآید که این بیماری زود شناخته شده باشد. حدس در زندهماندن افراد مبتلا به این بیماری اگر درمان Immunochemotherapy را نگرفته باشند، کمتر از ۱۰درصد است.
بهطور کلی HLH یک سندرم پاتولوژیک است که در آن فعالیت ایمنی پاتولوژیک با تظاهرات شدید التهابی (Inflammation) همراه است.
HLH ابتدا بهصورت عارضه ارثی در سال ۱۹۵۲ تحتعنوان Familial Hemophagociytic Reticulosis شناخته شد و آن هنگامی بود که دو برادر و خواهر بیمار در سنین طفولیتدچارتبو Cytopenias، هپاتواسپلنومگالی و Coagulopathy شده بودند.
جالب اینجاست که به کودک دوم هورمونAdrenocorticotropin تجویز کردند و به بهبودی نسبی دست یافتند. این بیمار(She) ۹۴روز بعد از شروع نشانهها زنده ماند، اما برادرش تنها۲۱ روز زنده بود.
در اتوپسی هر دو این کودکان، Hemophagocytosis در عقدههای لنفاوی و طحال وجود داشت. اینها موارد اولیه HLH ارثی کلاسیک بودهاند. این دسته کودکانی که درگذشته سالم بودهاند، با ظهور نشانههای غیراختصاصی انفلاماسیون ناگهان دچار بیماریهای وخیم با نارسایی سیستمهای مختلف بدن میگردند. در کودکان این تشخیص بسیار چالشبرانگیز است زیرا صفات شبیه و حالات مشترک با عفونتها (Common condition) را دارند.
در تجربهای مروری در تگزاس درTexas Children Hospital بیشتر بیمارانی که در نهایت با آزمایش ژنتیک بهتشخیص رسیدند، اغلب تشخیص Kawasaki Disease داشتند.
چالش تشخیصی در کودکان بزرگتر و افراد بالغ بیشتر است (Magnified) زیراکه کمتر بهاین بیماری فکر میشود و بیماریهای فراوانتری در تشخیص افتراقی وجود دارند.
خصوصیات تشخیصی:
خصـــوصیات تشخیصــی در اجـــماع Histiocytic society clinical Trials HLH-94 در جدول(1)بهنظر میرسد.
گرچه مطالب این جدول کمک بزرگی در تشخیص مینماید ولی Specifity و Sensitivity آنها اعتبار Prospectlive را دارا نمیباشد، مضافاً این که خصوصیات بیشتر HLH مانند نارسایی پیشرونده کبد، Disseminated intravascular coagulation و یا آنسفالیت در آن گنجانده نشده است. صرفنظر از این موضوع، مطالب داخل جدول برای تشخیص مفید است.

HLH اولیه و ثانویه:
وقتی در مورد HLH صحبت میکنیم، باید مشخص نماییم که منظورمان اولیه و یا ثانویه است.
اولیه، مخصوص کودکانی است که بهطور ارثی مستعد فقر ایمنی (Immune deficiencies)میباشند.
ثانویه یا اکتسابی بودن مربوط به بیماران مسنتری میباشد که در آنها Significant Immune activity ممکن است در اثر آنتیژنهای مختلف شامل بیماریهای اتوایمیون، عفونتهای مقاوم، بدخیمیها و یا از دستدادن Inhibitory immune mechanism ایجادگردد.
بررسی بالینی سیستمیک برای بیماران مبتلا بهHLH اساسی و واجب است، بخصوص در بالغین با تشخیص جدید در آنهایی که عفونت مخفی(Occult)، سرطان و یا بیماری اتوایمیون وجود داشته باشد.

حتی در بیمارانی که تشخیص HLH مادرزادی با آزمایش ژنتیک دارند، تحریک عفونی موجب ظهور نشانههای بیماری میشود. بنابراین وجود عفونت همراه (Coincident)، بیماری ارثی را از اکتسابی افتراق نمیدهد.
بنابر نظر اجماع HLH-94 پیشآگهی هر دو نوع بیماری یکسان است. به غیر از بیماری اتوایمیون و سرطانی عامل HLH که در آن بررسی اصلی و درمان، برای بیماران مشابه است. کشف وجود عیب ایمنی خاص (Fixed) و خطر عود بیماری از نظر درمان، در ادامه درمان و یا پیوند مغز استخوان اهمیت دارد. از نظر پاتولوژی، هموفاگوستیک لنفوهیستیوز(HLH)با پرولیفراسیون و فعالشدن هیستیوسیتها با نشانه فعالیت هموفاگوسیتوز مشخص میشود.
هموفاگوسیتوز همراه زیربنای عارضه ژنتیک و یا ثانویه با زیربنای عفونی، اتوایمیون یا پروسه نئوپلاستیک میباشد. مکانیسم پاتوفیزیولوژیک زیربنای اولیه HLH بهنظر میآید مربوط به آنومالی Cytokine باشد که موجب تجمع غیرقابل کنترل T-lymphocytes و Histiocytes میگردد.
نشانههای اولیه HLH ممکن است شبیه عفونت سیستمیک، هپاتیت یا آنسفالیت باشد. شایعترین نشانههای بالینی شامل تب، هپاتومگالی (حدود۹۰درصد)، اسپنومگالی۸۰درصد، سیستم عصبی ۴۵درصد و لنفادنوپاتی ۴۰درصد میباشد. پیشآگهی بالینی خراب بوده و تأخیر در درمان ممکن است سبب نارسایی غیرقابل جبران ارگانها و یا مرگ گردد. درمان توصیه شده برپایه راهنمای پروتکل HLH-94 شامل درمان حملهای (Induction) با دگزامتازون و Etoposide و سپسCyclosporineو Dexamethasone میباشد.
با این پروتکل که طول زمان زندگی ۳ساله ۵۵درصد گزارش شده، پیوند مغز استخوان بهترین انتخاب است.
چهار نوع ماژور هموفاگوسیتیک لنفوهیستیوز عبارتند از:
۱-HLH فامیلی.
۲ـ HLH توأم با عفونت.
۳ـ HLH توأم با بیماری اتوایمیون و عارضه Immune deficiency
۴ـ HLH توأم با سرطان.
Familial HLH یک عارضه Autosomal Recessive است که اطفال را از بدو تولد تا ۱۸ ماهگی در برمیگیرد و بهعلت موتاسیون ژنهایPRF1, LINCI3D و STX11 میباشد.
Infection- associated HLH: بهعلت طیف وسیع عفونتهای میکروبی و ویروسی، شامل Parvovirus, CMC, EBV, Varicella Zoster, Herpes simplex، سرخک، HIV، سل، عفونتهای گرم منفی، قارچی و انگلی گزارش شده است.
HLH associated with autoimmune و عارضه Immune deficiency که با SLE، ارتریت روماتوئید پلیارتریت Nodosa، سارکوئیدوز ریوی، سندرم Sjogren، نقص ایمنی، سندرم لنفوپرولیفراتیو X-linked بیماری Kawasaki و Chediak-Higashi همراه است.
HLH وابسته به سرطان را بهطور عمده با T-cell lymphoid malignancy، همینطور NK-cell Leukemia و لنفوم B-cell گزارش کردهاند.
مرفولوژی و نشانههای آزمایشگاهی:
HLH معمولاً عارضه سیستمیکی است که تمایل به گرفتاری در مغز استخوان، طحال، کبد و عقدههای لنفاوی دارد. سایر مناطق شامل مننژ، ریه، دستگاه گوارش، تیموس و دستگاه تناسلی ادراری نیز گرفتار میشوند. ولی پوست بهندرت دچار میگردد.
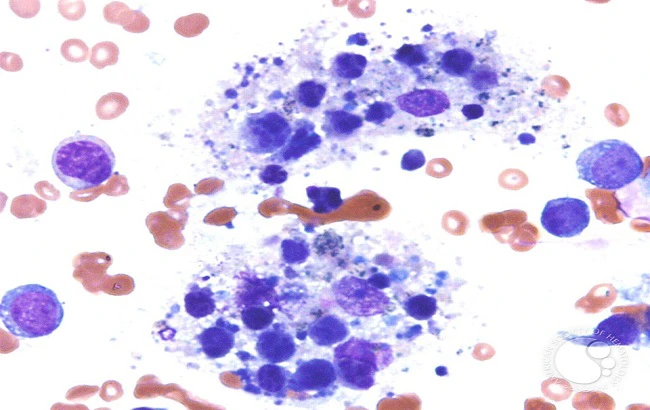
در نسج گرفتار وجود مختلطی از لنفوسیت، هستیوسیت بهصورت موضعی و Sinusoidal منتشر دیده میشود.هیستیوسیتها گرانولهای ظریف فراوان، سیتوپلاسم واکونوله، هستهگرد، بیضی شکل یا شیاردار (Cleaved) و هستک نامشخص دارند. هستیوسیتها بهتعداد زیاد وجود دارند که اغلب هموفاگوسیت گلبول قرمز و سایر سلولهای خونی مانند گلبولهای سفید یا پلاکتها در داخل آنها ملاحظه میشوند. Mitotic figures یا وجود ندارد و یا خیلی نادر است.تصاویر (۱و۲و۳)


آزمایش مغز استخوان بهترین وسیله برای تشخیص بیماری است ولی در بعضی از موارد تکرار آزماش مغز استخوان برای نشاندادن Hematophagocytosis لزوم پیدا میکند.
یافتههای دیگر بیماری عبارتند ازCytopenia،افزایش تریگلیسریدخون، هیپوفیبرینوژنمی، افزایش Ferritin خون، فقدان فعالیت NK-cell و افزایش IFN-d، فاکتور نکروز تومور(T.N.F) و اینترلوکینهای ۱۸،۱۲،۱۰،۶،۲،۱.در رنگآمیزی ایمیونوفنوتایپ و سایتوکمیکال، هیستیوسیتها تجلی CD68 و لیزوزم، CD10 منفی، اسیدفسفاتاز و Alpha1-antitrypsin مثبت را نشان خواهند داد.
در بررسیمولکولر و سایتوژنتیک، موتاسیون ژنهای LINC13D و STX11 اغلب در نوع فامیلیال گزارش شده است.
تشخیصهای افتراقی:
HLH از نظر بالینی ممکن است با سندرم نارسایی ارگانهای متعدد شامل ریوی، کاردیوواسکولار، کلیه و کبد شباهت داشته باشد. گرفتاری سیستم مرکزی اعصاب مشابه با آنسفالیت و پانسیتوپنی همانند نارسایی مغز استخوان و لوسمی میباشد. بیشتر موارد HLH توأم با زیربنای عفونت، عارضه اتوایمیون و بدخیمیهای لنفوئیدی میباشد.
تظاهرات بالینی و وجود Hemophagocylic histiocytes آن را از عوارض دیگرچونLysosomal storage وLangerhans cell histiocytes مجزا مینماید.
(جدول شماره۲): خصوصیات تشخیصی HLH را نشان میدهد.












