

دیستروفیهای عضلانی گروهی از بیماریها هستند که با 4 کرایتریای ضروری از سایر بیماریهای نوروموسکولر افتراق داده میشوند. این کرایتریاها عبارتنداز:
1ـ میوپاتی اولیه
2ـ انتقال ژنتیک
3ـ سیر پیشرونده
4ـ دژنرسانس فیبرهای عضلانی در زمانی از سیر بیماری.
دیستروفیهای عضلانی گروهی از اختلالات ارثی با درجات متغیری ازمحل انتشار ضعف عضلانی هستند. حد اقل 8 نوع اصلی دیستروفی عضلانی وجود دارد که شامل موارد Distal,Congenital ,Oculopharyngeal,Limb girdle,Facioscapulohumeral, Emery Dreifuss,Becker, Duchenne میباشد.
دیستروفی عضلانی مادرزادی:
گروه هتروژنی از اختلالات ارثی با توارث اتوزومال مغلوب هستند که در ابتدای تولد و یا چندماه اول تولد دچار ضعف عضلات پروکزیمال و هیپوتونی میشوند که به آهستگی پیشرفتکرده و با یافتهای دیستروفیک در عضله همراه است. نقایص ساختمانی مغزبا یا بدون MR در مواردی دیده میشود.
بهطورتقریب40 درصد شیرخواران با CMD کمبود اولیه زنجیره آلفا دو Laminin مروزین دارند که بهعلت موتاسیون در ژن LAMA 2 است. ایــن فرم را CMD1A نامند. جایگاه ژنی این فرم6q22-23 است. ضعف عمومی، دیسفاژی و گاه آپنه دیده میشود.
در فرم فوکویاما تشنج و MR شدید دیده میشود که حاصل نقص در Fukutin با جایگاه ژنی 9q31-33 است. این فرم از CMD در ژاپن بعداز دوشن شایعترین است. فوکوتین یکیاز گلیکوپروتیینهای همراه با دیستروفین است که در مغز و عضله اثر دارد. ضعف عضلانی شدید از هنگام تولد با بروز کونتراکچر زودرس و تظاهرات سیستم عصبی مرکزی نشانگر این بیماری است. کاهش شدید در دانسیتهی ماده سفید و آنومالیهای مهاجرت نورونها از یافتههای تصویربرداری مغز دراین بیماری است. بیماری در انتهای دههی اول منجر به مرگ میشود.
دیگر از انواع CMD براساس وجود و یا فقدان مروزین به دودسته تقسیم میشوند. مروزین زنجیرهی آلفا 2 لامینین است که به سارکولما متصل میشود. درصورت فقدان مروزین لامینای بازال ناپدید و سارکولما دژنرهشده و سلول عضلانی نکروز میشود. مروزین در پوست، سلولشوان و مغز وجود دارد. مروزین را میتوان با ایمونوهیستو کمیستری در بیوپسی فروزن عضله نشانداد. کاهش مروزین در CMD1A Muscle-Eye-Brain Disease , CMD1B و فرم فوکویاما دیده میشود.
یکی از انواع مروزین مثبت CMD سندرم Walker-Warburg است. دراین سندرم لیزنسفالی نوع2، کیست دندی واکر، ناهنجاری مخچه و رتین دیده میشود. نقص در ژن POMT1 دراین سندرم مطرح است.
فرمی از CMD بهنام Muscle-Eye-Brain Disease.حاصل نقص در ژن POMGnT1 است . هیپوتونی مادرزادی، میوپی و MR از یافتههای بالینی این بیماری است و بیشتر در فنلاند دیده میشود.
یکیاز انواع مروزین مثبت CMD بیماری Ullrich است. در این بیماری ضعف عضلانی پیشرفت آهسته دارد و با کونتراکچر مفاصل پرکزیمال و هیپرموبیلیتی مفاصل دیستا ل همراه است. این بیماران از هوش طبیعی برخوردار هستند. تعریق زیاد و برجستگی پاشنه پا کاراکتریستیک بیماری است.
بیماری دوشن و بکر (دیستروفینوپاتی):
دیستروفین۰/۰۰۲ کل پروتئینهای عضله را تشکیل میدهد و درسطح داخلی سارکولما قراردارد. کمپلکس دیستروفین، گلیکوپروتئین شامل چندین گلیکوپروتئین همراه با
دیستروفین و پروتیین همراه با دیستروفین است.
دیستروفین علاوهبر عضله در مغز و رتین هم وجود دارد.
دیستروفین در محل سارکولما با دیگر پروتئینها همراه است ازجمله آلفا و بتا دیستروگلیکان و یوتروفین.
معمولاً در دوشندیستروفین وجود نداشته و در بکر کاهش داشته و یا غیرطبیعی است. ژن این دو بیماری در جایگاه Xq21.2 است. شایعترین موتاسیون در این ژن Deletion است.
دیستروفی عضلانی دوشن در سال1868 توسط دوشن و در سال1879 توسط Gowers مطرح شد. شیوع بیماری1 در25000 است. بیماری در اوایل دوران کودکی با مشکل در با لارفتن از پلهها بارز میشود و بهتدریج برشدت ضعف افزوده شده و راهرفتن اردکی Waddling Gait و Gowers,sign دیده میشود. هیپرتروفی کاذب ساق پا و کونتراکچر شایع است.
بزرگی قلب، تاکیکاردی و نارسایی قلب دربین50 تا80 درصد بیماران دیده میشود.
اختلالات عضلات صاف بصورت اتساع شکم، استفراغ، دردشکم و یبوست وجود دارد.
درصد قابلتوجهی از بیماران از ضریب هوشی کمتراز طبیعی برخوردار هستند و متوسط ضریب هوشی بیماران 85 است. اختلال در ساختمان کورتکس و دندریتهای نورونهای که دیستروفین دارند علت مشکلات هوشی بیماران است.
سیر بیماری پیشرونده و بیمار را زمینگیر میکند. عفونت و نارسای قلب در دوران نوجوانی باعث مرگ میشود.
ناقلان دختر معمولاً بدونعلامت هستند ولیکن گاهی هیپرتروفی کاذب و مختصر ضعف عضلات دارند.
دیستروفی عضلانی بکر از شیوع کمتری برخوردار است. بیماری معمولاً بعد از 8 سالگی بارز میشود. میزان هیپرتروفی کاذب، MR و درگیری قلب از دوشن کمتر است ولیـــکن در60 درصد مـــوارد pes cavus دیده میشود.
دیستروفی عضلانی محور اندامها: Limb Girdle Muscular Dystrophy
شامل گروه هتروژنی از حالات بالینی است. فرم با انتقال توارث مغلوب شایعتر است و بهنام LGMD2 میباشد. حداقل10 جایگاه ژنی برای این فرم شناخته شدهاست. فرم با توارث غالب به نام LGMD1 و با 6 جایگاه ژنی وجود دارد.
تظاهرات بیماری در دههی دوم یا سوم بارز میشود و در ابتدا محور شانه یا لگن را گرفتار میکند. سیر بیماری متغیر بوده ولیکن از دوشن خوشخیمتر است. در 30 درصد از موارد هیپرتروفی کاذب بهخصوص در ساق پا ملاحظه میشود. میزان هوش در بیماران طبیعی و درگیری قلب نادر است مگر در نوع1B. یکیاز انواع شایع با توارث مغلوب نوع1C است که حاصل موتاسیون در Caveolin 3 است.
انواع شایع با توارث مغلوب فرم 2A و 2B است. فرم 2A حاصل نقص در ژن 3 -Calpain است. این فرم از حوالی10سالگی تظاهر داشته و با MR و درگیری قلب همراه نیست.
LGMD2B حاصل نقص در ژن Dysferlin است که با دیستال میوپاتی شباهت دارد.
خیلی از انواع با توارث مغلوب حاصل نقص در یکی از 4نوع سارکوگلیکان است. نقص در سارکوگلیکان آلفا یا ادهالن باعث LGMD2D میشود که از شایعترین انواع است.
تشخیص آزمایشگاهی:
1ـ اندازهگیری کراتین کیناز که در دوشن حتی از هنگام تولد افزایش نشان میدهد.
2ـ الکترومیوگرافی که میتواند ضمن رد علل نوروژنیک پدیده میوژنیک را اثبات نماید.
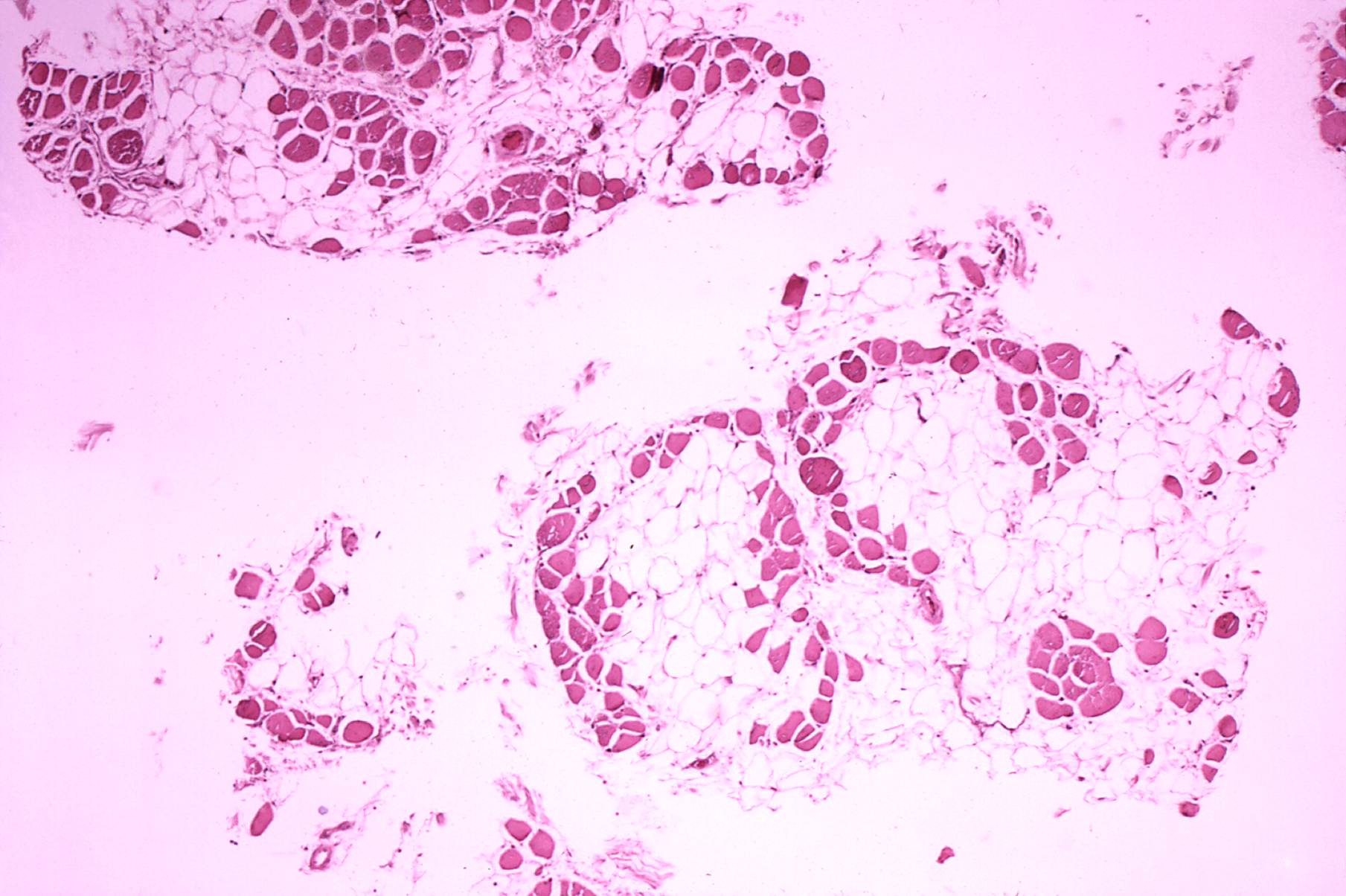
3ـ نمونهبرداری از عضله که نشانگر نکروز، تفاوت در اندازهی فیبرهای عضلانی، تهاجم ماکروفاژها و درنهایت وجود چربی است.
4ـ ایمونوهیستوکمیستری با استفاده از آنتیبادیهای نشاندار برعلیه پروتئینهای سارکولما اساس تشخیص است.
ثبت نظر