

دژنراسیون لوبپیشانی گیجگاهی(FTLD) بیماری تخریبکنندهی نورون است که معمولاً سبب زوالعقل میشود.
FTLD در کار بالینی، یک سندرم بهحساب میآید و درحالحاضر بهواسطهی معیارهای جمعی (Consensus Criteria) گروهی از محققان به ۳زیرگونه تقسیم شدهاست: دمانس پیشانی گیجگاهی (FTD)، آفازی ناروان پیش رونده و دمانس معنایی. بیشتر مبتلایان به آفازی (Nonfluent) پیـشرونده و دمانس معنایی (Semantic dementia). بعدها در دورهی بیماری خود برخی ویژگیهای FTD (مثل نشانههای رفتاری) را ازخود نشانمیدهند.
گفتهمیشود مبتلایان به FTD که تصویر بالینی برجستهی مرحلهی آغازین بیماری است، مبتلا به فراموشی پیشانیگیجگاهی گونهی رفتاری(bvFTD) هستند.
نوروپاتولوژی FTLD بههمان پیچیدگی سندرم بالینی است. در واقع، مولکولهای خاص بیماری در سلولهای تمامی مبتلایان به FTLD بهشکلی غیرعادی انباشته شدهاند. این مولکولها شامل Tau، پروتئیـن۴۳ متصـلشونده به (TAR DNA (TDP-۴۳ و سارکوم الحاقشده(FUS) است. موارد FTLD اکنون برای یکیاز ۳زیرگروه مولکولی اصلی تعیین شده است که بر مبنای یافتههای هیستوپاتولوژیکال میباشند. این ۳زیرگروه مولوکولی اصلی عبارتنداز: FTLD-TDP وFTLD-FUS. قبلاز کشف TDP-۴۳ درسال۲۰۰۶، بیشتر موارد FTLD دارای Tau منفی، مجموعاًFTLD-U نامیده میشدند؛ زیرا انکلوزیون یوبیکویتین مثبت (Ubiquitin Positive) بودند.
متعاقباً آشکارشد که بیشتر مواردFTLD-U درواقعFTLD-TDP (یعنی انکلوزیونهایFTLD همراه با TDP-۴۳) بودند و ۱۰ تا ۲۰ درصد از موارد FTLD-U بهشکلFTLD دارای Tau منفی و FTLD دارای TDP-43 منفی باقیماندند. درسال ۲۰۰۹، FUS شناساییشد که یکیاز ژنهای بیماری خانوادگی اسکلروز جانبی آمیوتروفیک (ALS) بود. ازاینرو، مشخصشد که تمامی گنجیدگیهای FTLD تائو منفی و TDP-۴۳ منفی FUS مثبت بودند. بنابراین گنجیدگیهای FTLD دارای FUS مثبت اکنون مجموعاً FTLD-FUS نامیده میشوند. ۳شکل نادر FTLD از زیرگونههایFTLD-FUS بهحساب میآیند. این ۳نوع نادر عبارتنداز: FTLD-U غیرعادی (a-FTLD-U)، بیماریBIBD و بیماری نورونی گنجیدگی فیلامان واسط (NIFID: Neuronal Intormediate Filament Inclusiun Disease). هرچند این ۳زیرگونه ممکناست طیفی پیوسته از بیماری FTLD-FUS باشند، بررسیهای تفصیلی هیستوپاتولوژیک نشانمیدهد که در عین داشتن رابطهای تنگاتنگ، موجودیتهایی مستقل میباشند.
بسیاریاز گزارشهای سابق، این روابط کلینیکوپاتولوژیک را در بیمارانFTLD به چالش کشاندهاند. در FTLD-FUS که در تعداد کمی از مبتلایان به FTLD وجود دارد، این روابط تنها همین اواخر تشریح شدهاند. این تحقیقات نشانمیدهند که مبتلایان بهFTLD-FUS، احتمالاً شروع بیماری به نسبت کمتری (اغلب قبلاز ۴۰ سالگی) دارند، سابقهی خانوادگی ندارند و در عکسبرداریها، آتروفی دمی شدیدی نشاندادهاند. اخیراً، اسنودن و همکاران نشاندادهاند که a-FTLD-U با فنوتیپی شناختی و رفتاری مرتبط است. این فنوتیپ شناختی و رفتاری از دیگر اشکال FTLD-FUS (بخصوص NIFID و (BIBD: Basophilic Inclusion Body Disease) متمایز است.
علاوهبراین، محققان بیان میدارند؛ نتایج بالینی پاتولوژی FTLD-FUS ممکناست با جهش ژن FUS ارتباطی نداشته باشد. این افراد میگویند که مشخصات a-FTLD-U ممکناست وسواسی بودن زیاد، رفتارها و مراسم تکراری، کنارهگیری از اجتماع و بیکاری، هایپراورالیتی همراه با هرزهخواری و رفتار آشکار وابسته به محرک (مثل رفتار مصرف) باشد. بهعلاوه در تعداد کمیاز تحقیقات، گزارش شدهاست کهBIBD وNIFID دارای فنوتیپ بالینی یکپارچهای هستند. یوکودا و همکاران دریافتند که NIFID و BIBD دارای ویژگیهای کلینیکی مشترک بسیاری ازجمله دیسآرتری، نشانههای نورونهای حرکتی، پارکینسون و اختلال حافظه هستند. این افراد همچنین خاطرنشان کردند که متمایزکردن BIBD از NIFID در روشهای بالینی کاری دشوار است. درحالیکه این گزارشها تغییراتی را در ویژگیهای رفتاری و شناختی FTLD-FUS نشانمیدهند، ویژگیهایی که متمایز از دیگر اشکالFTLD (یعنیFTLD-Tau و FTLD-TDP) هستند باید مشخص گردند. بیماران مبتلا به FTLD اغلب میگویند که دارای اختلالات مرتبط با سیستم حرکتی همچون پارکینسون و بیماری نورونهای حرکتی هستند، درحالیکه بهندرت از ارتباطFTLD با تشنج و آتتوز گفتهاند. در معیارهای تشخیصی بالینیFTLD، کوریاآتتوز یکیاز ویژگیهای تشخیصی استثناء است. تشنج، بیماری حرکات غیرارادی و غیرعادی با ویژگی حرکات بیشازاندازه و غیرارادی است. این حرکات زمان بیقاعدهای دارند و غیرتکراری، تصادفی و ناگهانی میباشند. شکل کلاسیک بیماری تشنج در بیماری هانتینگتون (HD) روی میدهد. بیماری هانتینگتون یک بیماری ارثی فاسدکنندهی نورونها است که درآن آتروفی استریاتوم، پاتولوژی غالب است. بااینحال، استریاتوم نیز شدیداً در همهی انواع فرعیFTLD تحتتأثیر است. نتایج اخیر MRI مبتلایان به FTLD-FUS بهطور معناداری نشانداد که آتروفی استریاتال آنها نسبت به مبتلایان به FTLD-TDP و FTLD-Tau بیشتر است. درنتیجه، میتوان FTLD-FUS را از انواع دیگر FTLD تشخیصداد.
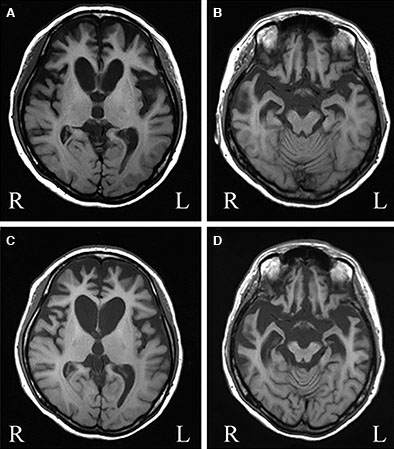
شکل۱. MRI مغزی مورد 1. الف و ب. MRI مغزی بیمار مورد ۱ در ۳۶ سالگی. در قشرهای پیشانی و گیجگاهی و هستۀ دمی، آتروفی وجود دارد. MRI مغزی A و B این بیمار ۱سال پساز C و D گرفتهشدهاند. آتروفی شدیدتر و شاخ قدامی بطنهای طرفی مشخصاً بزرگ شده اند.
دراینجا، برای بیماری تشنج ۳مورد bvFTD را شناساییکردهایم. این ۳ مورد FTLD-FUS تشخیص دادهشدهاند و نتایج هیستوپاتولوژیکی را نشانمیدهند که نشانهای از نوع فرعیBIBD هستند. ویژگیهای رفتاری و شناختیای را شناسایی کردهایم که این گروه را از دیگر موارد FTLD-FUS متمایز میسازد. بهعلاوه، بهمنظور شناسایی ویژگیهای متمایز بالینی که نشاندهندهی FTLD-FUS و مخصوصاً نوع فرعی BIBD هستند، سوابق بالینی ۷۲موردFTLD را نیز بررسیکردهایم.
نتایج یافتههای بالینی:
مورد اول:
بیمار زنی ژاپنی با سابقهی خانوادگی شیزوفرنی بود اما سابقهی خانوادگی دمانس یا اختلالات حرکتی نداشت. وقتی در ۳۲سالگی مبتلا به بیاختیاری شد، سالهای زیادی را خارج از ژاپن زندگیکرده بود. ۲سال بعد، بیشتر زمان روز را در رختخواب میگذراند، مبتلا به پالیلالیا بود و نسبت به کربوهیدراتهایی ازقبل برنج و رشتهفرنگی اشتهای زیادی داشت. بهشکلی تحریکآمیز لباس میپوشید و اغلب بهخاطر دزدی از مغازهها بازداشت میشد، اما اظهار پشیمانی نمیکرد. بیماری او در ابتدا شیزوفرنی یا اختلال افسردهکننده تشخیصداده شد و درنتیجه، با فلوواکسامین و اولانزاپین و نیز با ECT تحتدرمان قرارگرفت.
این زن به درمانشهایش علاقهای نداشت و درنتیجه، درمانها متوقفشدند. در ۳۶سالگی به ژاپن بازگشت و در بیمارستانی روانی بستری شد. درصورت، زبان، گردن و دست و پاهایش حرکات غیرارادی تشنج مانندی وجود داشت. این حرکات غیرارادی شامل انقباض مکرر شانهها، حرکات پیوستهی عضلات صورت (مثل بالا بردن ابروها، بستن چشمها، بیرون آوردن زبان) و حرکات پردامنهی پاها، گهگاه با خشونت، پرتابکردن و کوبیدن بود که به آن بالیسیموس گفتهمیشود. همچنین در پای راستش، حرکات آتتوز مانندی وجود داشت. وقتی در حیاط بیمارستان میچرخید، چیزهای زرد را لمس میکرد و آهسته به آنها ضربهمیزد. معاینات نورولوژیکی نشاندهندهی کاهش تونوس ماهیچهای بود اما ضعف ماهیچهای، آتروفی یا دیگر نشانههای بیماری نورونهای حرکتی را نشاننمیداد. قدرت تکلمش کاهشیافته بود، اما برخیاز لغات ساده را بیانمیکرد. رفتارش قالبی و تشریفاتی بود. نتایج آزمایش بیوشیمی خون او ازجمله سطوح فریتین و سرولوپلاسمین و آزمایشهای سیفیلیس عادی بود.
در مایع مغزی ـ نخاعی غلظت پروتئین بتاآمیلوئید، مجموعTau وTau فسفریلشده عادی بودند. نتایجMRI مغز نشاندهندهی آتروفی دوطرفهی پیشرونده در قشرهای پیشانی، گیجگاهی و هستهی دمی بود (شکل۱).
تصاویر سیتیاسکن خروج تکفوتونی جریانخون مغزی نشانمیدهند که هیپرفیوژن در این مناطق آشکار است. نتایج بررسی الکترومیوگرافیک عادی بود. بررسی وضعیت روانی برمبنای (MMSE) وی ۱۸/۳۰ بود. بهدلیل تشنج و آتروفی شدید دمی که در تصویربرداری MRI به روشنی دیده میشد، به لحاظ بالینی مشکوک به بیماری هانتینگنون بود. بااینحال، ژنهایی که سبب بیماری هانتینگتون میشوند، یعنی آتروفی دنتاتوروبرال ـ پالائیدولوزیان نوع ۱۷ آتاکسیای نخاعی ـ مخچهای، در او گسترش نمییافت. در ژنهای Tau،TDP-43،FUS، گرانولین، پروتئین پیشروی آمیلوئید، پریسنیلین۱ یا پریسنیلین۲ هیچ جهشی یافت نشد. برمبنای یافتههای بالینی، این زن مبتلا به bvFTD تشخیصداده شد و برای درمان، میلناسیپران هیدروکلرید (۱۰۰mg) مصرف میکرد. در ۳۷سالگی و ۵سال پساز شروع بیماری، قدرت تکلمش کاهشیافته بود و دیسفاژی (اشکال در بلع) داشت. شدت حرکات خشن غیرارادیاش کمترشده بود، اما حرکات پای پیوسته و آرام او همچنان وجود داشت. درآنزمان، معاینات نولولوژیکی بهطور دوجانبهای وجود واکنشهایی ابتدایی همچون مکیدن و نیز واکنشهای چنگزدن قوی و پالمومنتال را نشانمیداد. این زن تا ۳۸سالگی در وضعیت نباتی مداومی بود. بازوی وی منقبض میشد، اما حرکات آتتوز مانند (همچون چرخش بیرونی و درونی جریان پیوسته، موجی و کند پای راست) تا پایان عمر با او بود. این بیمار در ۳۹سالگی براثر برنکوپنومونی درگذشت. مدت بیماری وی ۷سال بود.
مورد دوم:
بیمار دوم یک زن ژاپنی ۴۷ساله بود. این بیمار هیچ سابقهی خانوادگی دمانس یا تغییر رفتاری نداشت. در ۴۴سالگی مبتلا به بیتفاوتی و مهارگسیختگی بود.اشتهای این زن زیاد شده بود و درنتیجه وزنش بهسرعت بالا میرفت. در 46سالگی بهخاطر پرسهزدنهای بیهدف در یک بیمارستانروانی بستریشد. قدرت تکلم وی کاهشیافته بود و به زبانی ساده و کلیشهای حرفمیزد و درجاماندگی داشت.
فقط با جملات کوتاه و ساده سخنمیگفت، اما درک کلامیاش بینقص بود. معاینات نورولوژیکی نشانمیداد زبان بیمار دارای حرکات غیرارادی تشنجمانند تند و پیوستهای است، اما آتروفی عضله وجود نداشت. قبل از اینکه تشنج کند، دارویی برای وی تجویز نشد. ضریب هوش وکسلر بزرگسالان وی ۶۰ (ضریب هوش گفتاریاش ۷۲ و ضریب هوش عملکردیاش ۵۵) بود. نتایج بیوشیمی خون وی ازجمله آزمایش سیفیلیس عادی بود. اختلالحافظه یا موقعیت ناآگاهی در وی آشکار نبود. این بیمار از خود رفتارهای قالبی همچون پرتاب مکرر مقدار زیادی دستمال توالت به داخل توالت نشانمیداد. تشنج زبانی او تا ۴۷سالگی بتدریج کاهشیافت. مرگ ناگهانی وی براثر خفگی حین خوردن غذا درطی اقامت در بیمارستان بود. طول بیماری او ۳سال بود.
مورد سوم:
بیمار سوم یک زن ژاپنی ۶۷ ساله بود. این بیمار سابقهی خانوادگی دمانس نداشت، اما خواهرش براثر یک بیماری روانشناختی عمومی فوت شده بود. این زن در ۵۶ سالگی شروع به جمعآوری آشغال و کشهای لاستیکیکرده بود و تنها غذایش برنج و ترشی بود. در ۵۸سالگی بهدلیل مصرف زیاد الکل و ارتکاب اقدامات مجرمانهای همچون سرقت از مغازه، دریک بیمارستان روانی بستری شد. بهشدت دچار اختلال در حافظهی کوتاهمدت و موقعیت ناآگاهی بود. نتایج معاینات نورولوژیکی وضعیت غیرعادی را نشاننمیداد. نتایج بیوشیمی خون و ادرارش عادی بود. در تیمارستان کلیشهی رفتاری و کلامیاش بتدریج بهبود یافت. در ۶۰ سالگی، قادر به تکلم نبود و بتدریج علیلشد. دستها و پاهایش بهطور آشکاری منقبض میشد. در ۶۵سالگی و ۹سال پساز آغاز نشانههایبیماری، در گردن، بالاتنه و دست و پاهایش، حرکات غیراختیاری تشنج مانند خفیف و سریعی مشهود بود که بتدریج بدتر شد. در دست چپش نیز حرکات آتتوزمانند آشکاری مشاهده میشد. این حرکات تا پایانعمر با او بود و در ۶۷ سالگی براثر نارسایی قلبی جانسپرد. دراین مورد، قادر نبودیم تا دادههایی را درمورد سابقهی دارویی وی بهدستآوریم. مدت بیماری او ۱۲سال بود.
ثبت نظر